

企业档案
会员类型:会员

已获得易推广信誉 等级评定
171成长值
(0 -40)基础信誉积累,可浏览访问
(41-90)良好信誉积累,可接洽商谈
(91+ )优质信誉积累,可持续信赖
易推广会员:8年
工商认证 【已认证】

最后认证时间:
注册号: 【已认证】
法人代表: 【已认证】
企业类型:生产商 【已认证】
注册资金:人民币万 【已认证】
产品数:91228
参观次数:11305349
技术文章
抗体偶联药物 (ADC)――抗肿瘤细胞 | MedChemExpress
点击次数:1057 发布时间:2021/9/6 10:28:25
Antibody-drug Conjugate (ADC)是一类结合了化学疗法和免疫疗法的药物。这个概念*早于 100 多年前由德国科学家/医生 Paul Ehrlich 提出。ADC 由抗体 (Antibody)、细胞毒素分子 (ADC Cytotoxin, payload) 以及连接两者的连接子 (Linker) 组成。Paul Ehrlich 将 ADC 比喻成“魔法 bullet ”,因为它能在不伤害有机体 (毒副作用小) 的情况下特异性识别目标 (癌细胞)。ADC 毒素分子通过破坏 DNA、微管蛋白等从而阻止肿瘤细胞分裂,起到杀死细胞的作用。ADC 的准确靶向,都消除,简直媲美“ sniper ”。
一个成功的 ADC 分子,首先要保留单克隆抗体的选择性,同时能够释放足够高浓度的 payload,从而达到杀死肿瘤细胞的目的。我们先看下 ADC 发挥作用的步骤和机制。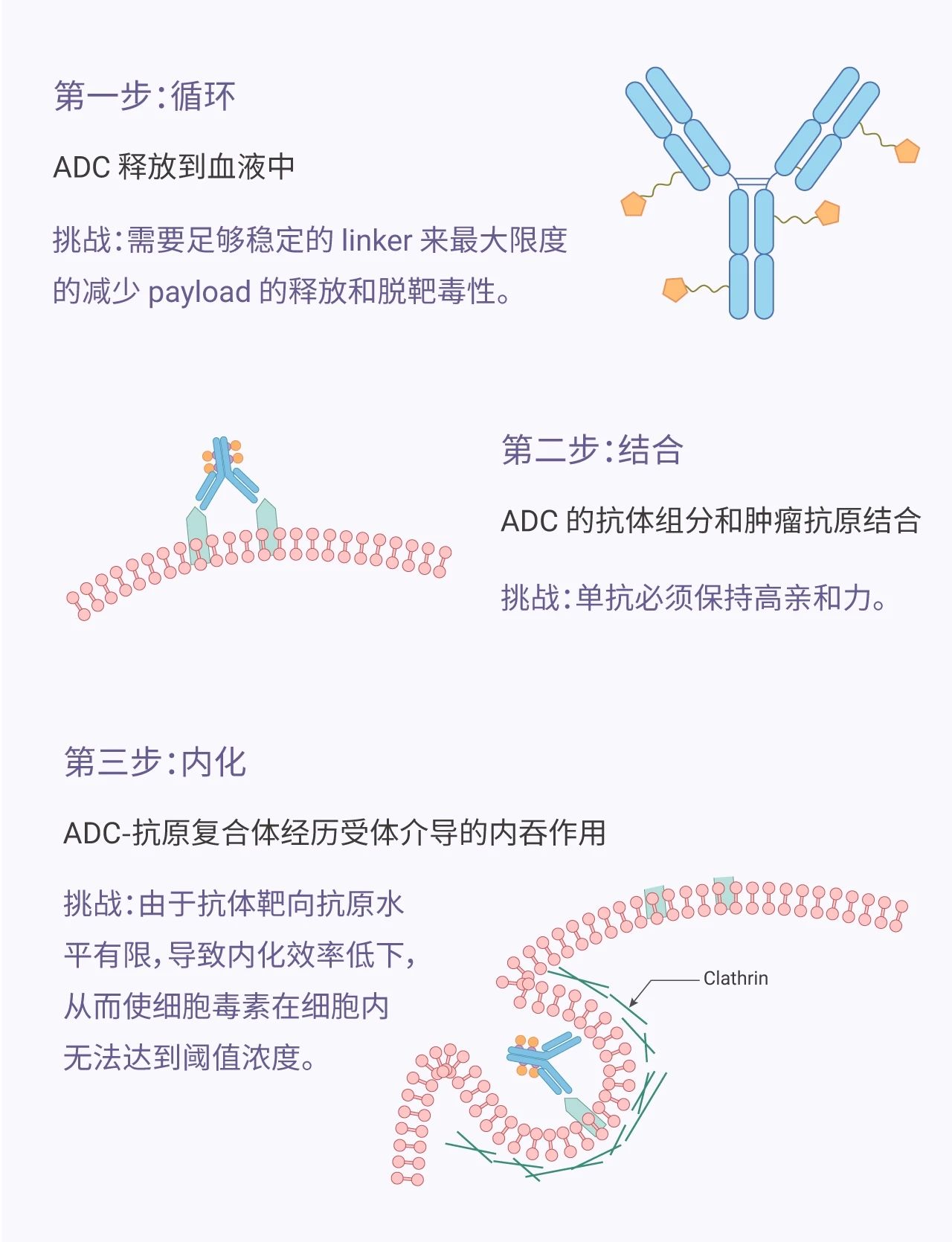
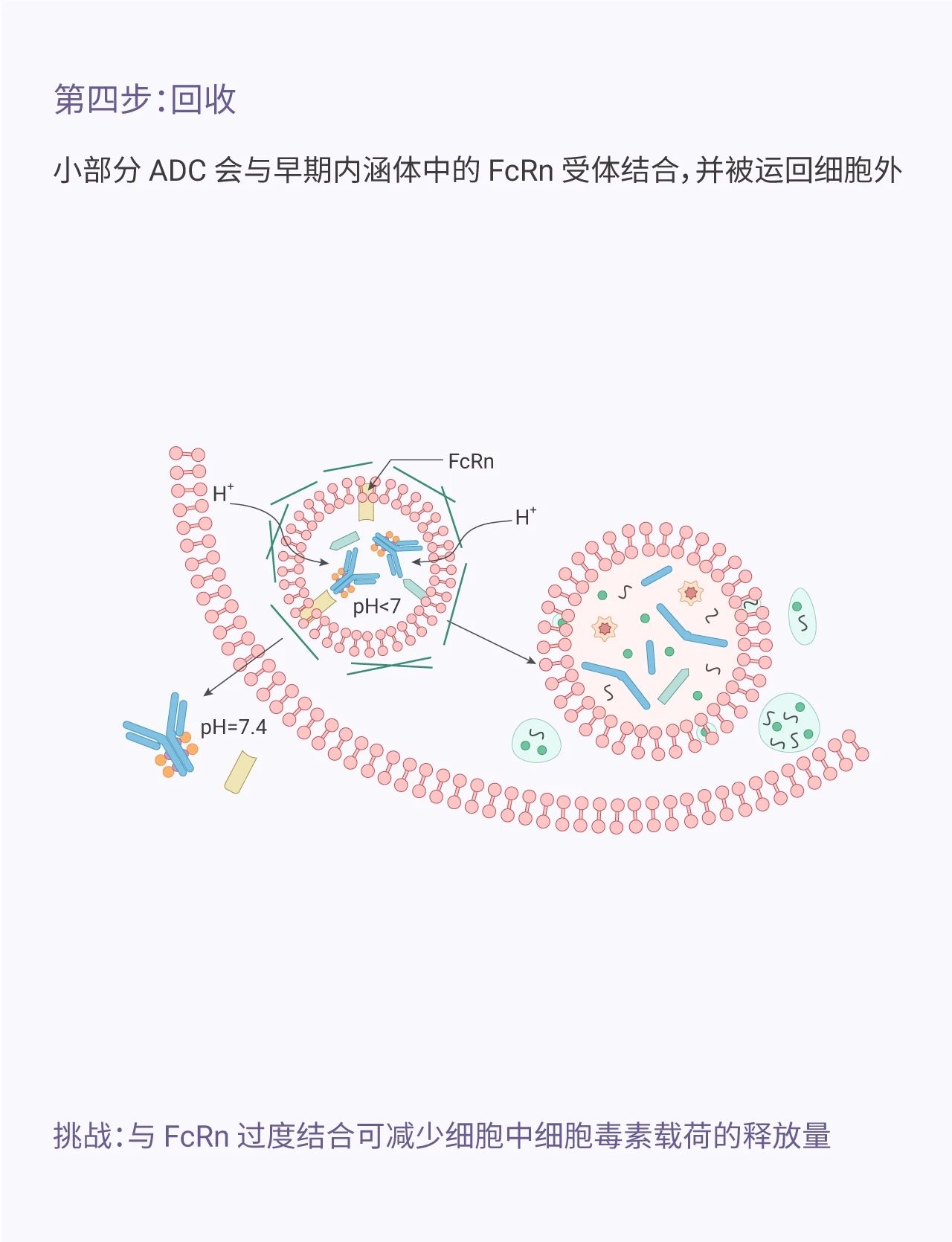
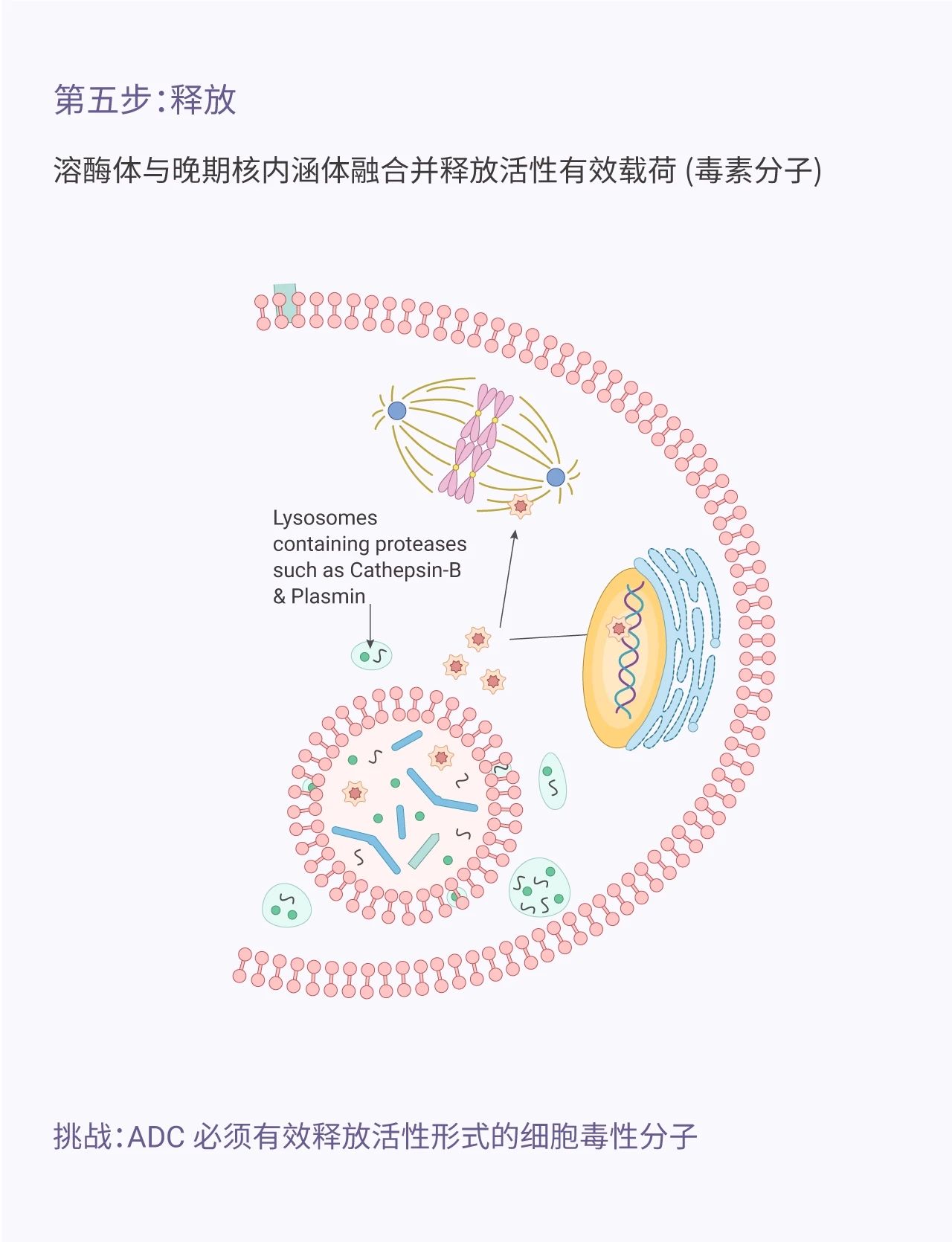
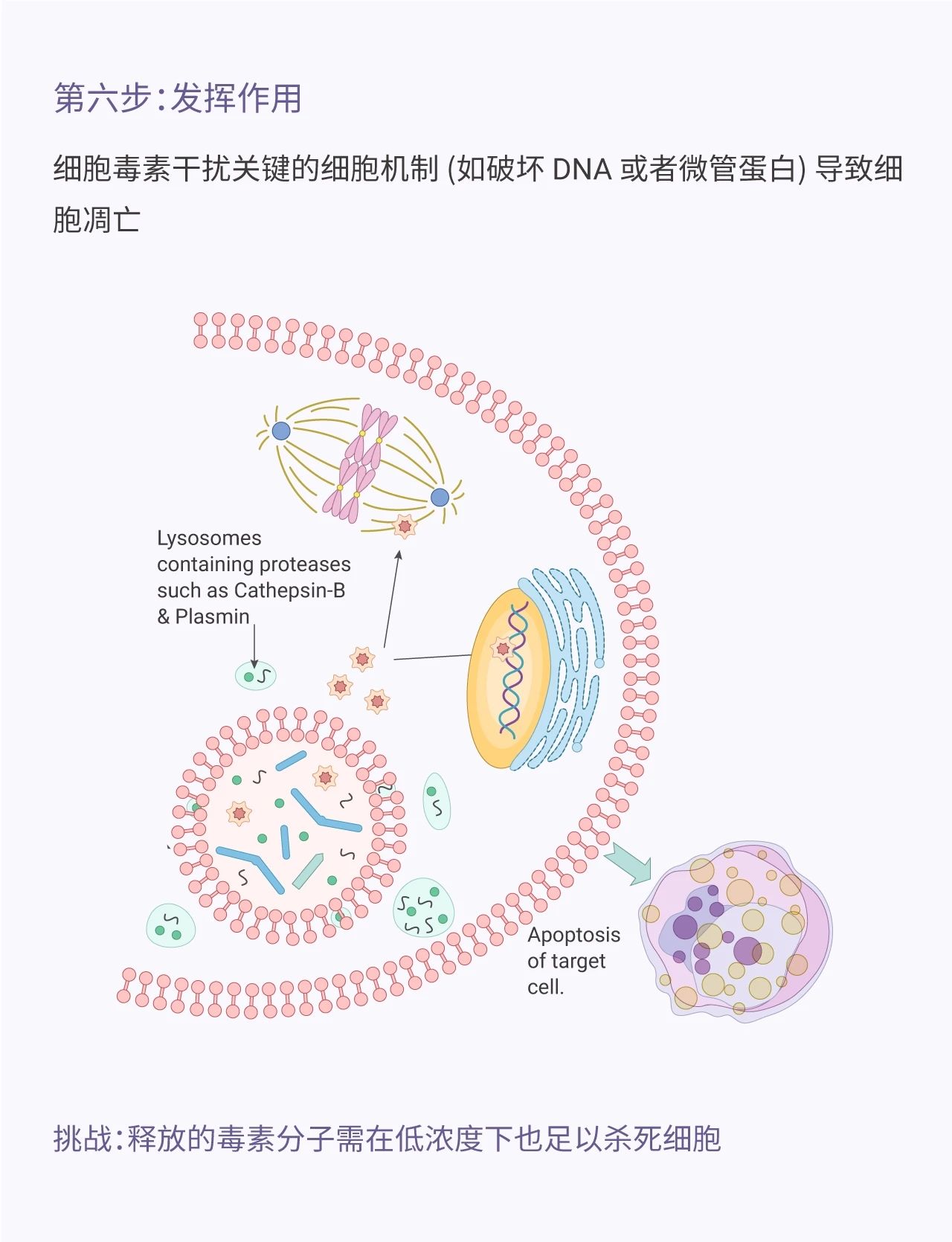
图 1. ADC 发挥作用的机制
综上,在一个成功的 ADC 分子中,抗体的特异性、linker 的稳定性以及 payload 的高活性缺一不可。
■ ADC 的各组分是如何组装成完整“魔法 bullet ”的呢?
抗体的连接位置集中在 IgG1 和 IgG4 家族。*早发展起来的方法是位于 linker-payload 末端的亲电子基团 (比如马来酰亚胺或 N-hydroxysuccinimide (NHS) 片段),与抗体暴露的赖氨酸残基组装在一起。这种“随机”偶联方法会得到 ADC 的异质混合物。这样的混合物会对药代动力学、耐受性和疗效等产生负面影响。直到后来开发出的位点特异性偶联方法,大大减少了异质性。
药物抗体比 (Drug-to-antibody ratio, DAR) 是抗体偶联药物的平均数量,是 ADC 的一个重要属性。目前的偶联化学方法是赖氨酸侧链酰胺化或半胱氨酸链间二硫键还原,通常每个抗体的药物负载为 0~8 个药物 (D0~D8)。
通过赖氨酸残基结合:虽然赖氨酸残基是有效的亲核基团,但是因为在整个抗体结构中分布着 80~100 个赖氨酸残基。所以作为*早开发的偶联方法,其选择性低。
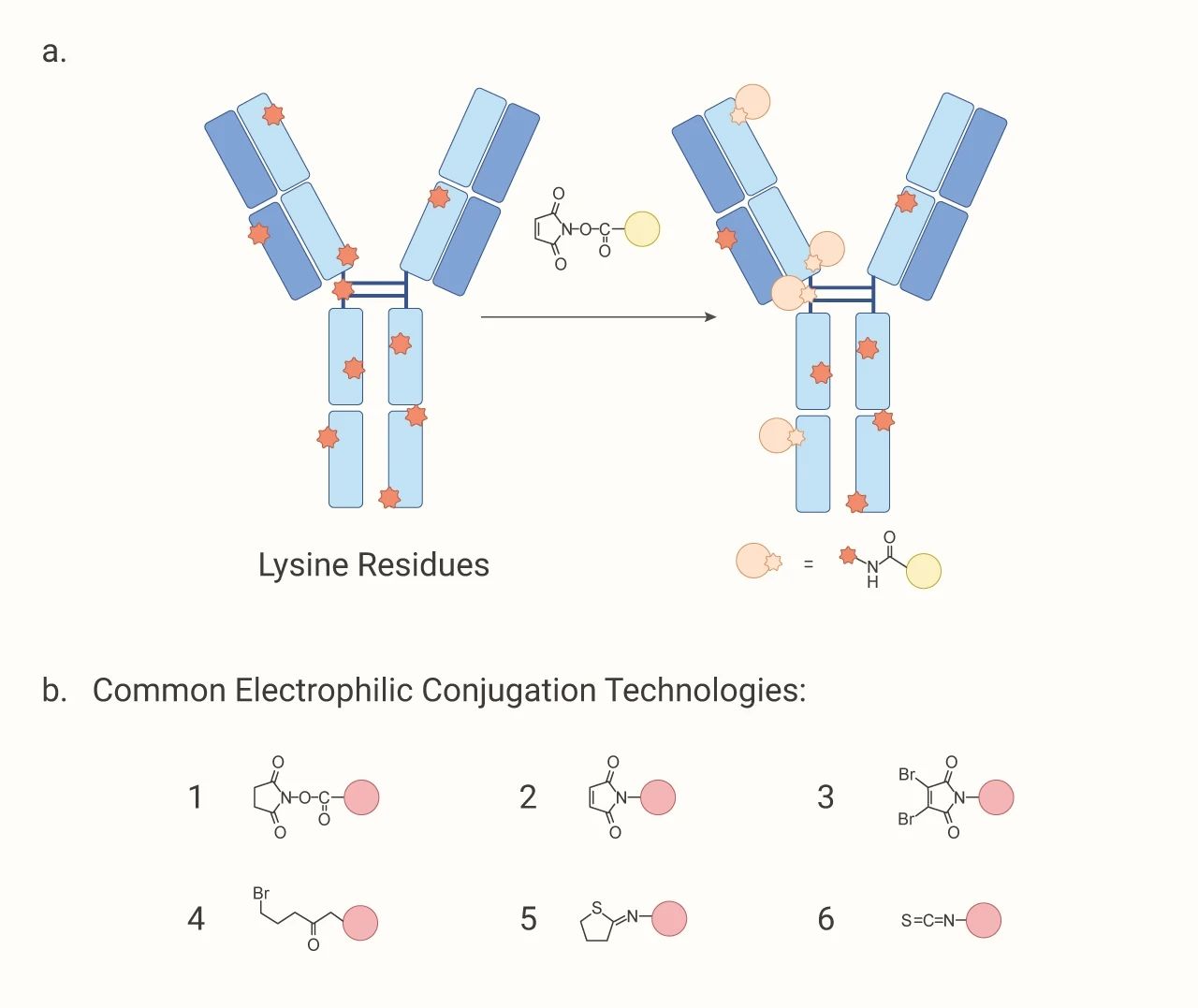
图 2. 赖氨酸残基结合的偶联过程
通过硫醇基团偶联:一般来说,抗体不含游离硫醇,目前*常用的是人类 IgG1 抗体,有 4 个链间二硫键和 12 个链内二硫键,传统方法需要用 TCEP、DTT 或者 2-MEA 还原剂将 4 个链间二硫键氧还原打开,产生游离的硫醇,与 linker-payload 复合体中的亲电基团发生反应 (马来酰亚胺、NHS),产生不同 DAR 数和不同连接位点的 ADC 偶联异质混合物。
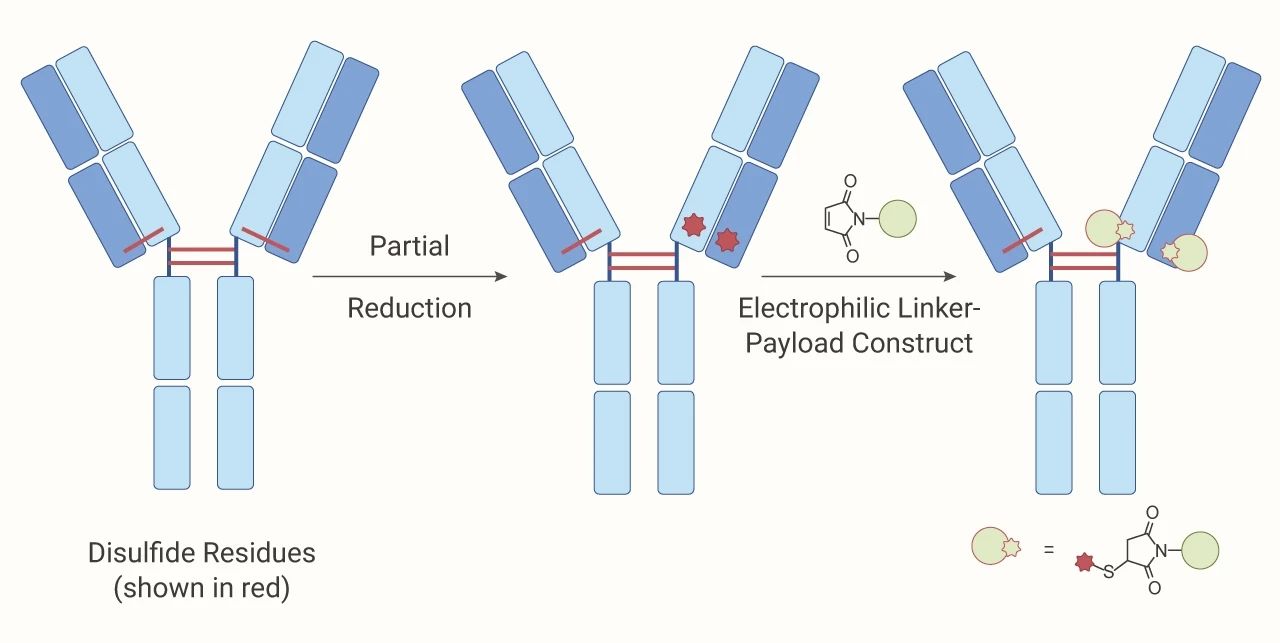
图 3. 硫醇基团偶联过程
位点特异性偶联 (硫醇基团的特异性偶联):*出名的技术要数 Genentech 的 THIOMABTM 技术了。THIOMABs 利用基因工程技术在抗体特定位置插入带有半胱氨酸残基 (游离硫醇)。半胱氨酸残基与 payload 偶联,这种定点偶联方式,得到的 ADC 的 DAR 为 2。在体内实验显示出好的安全性。
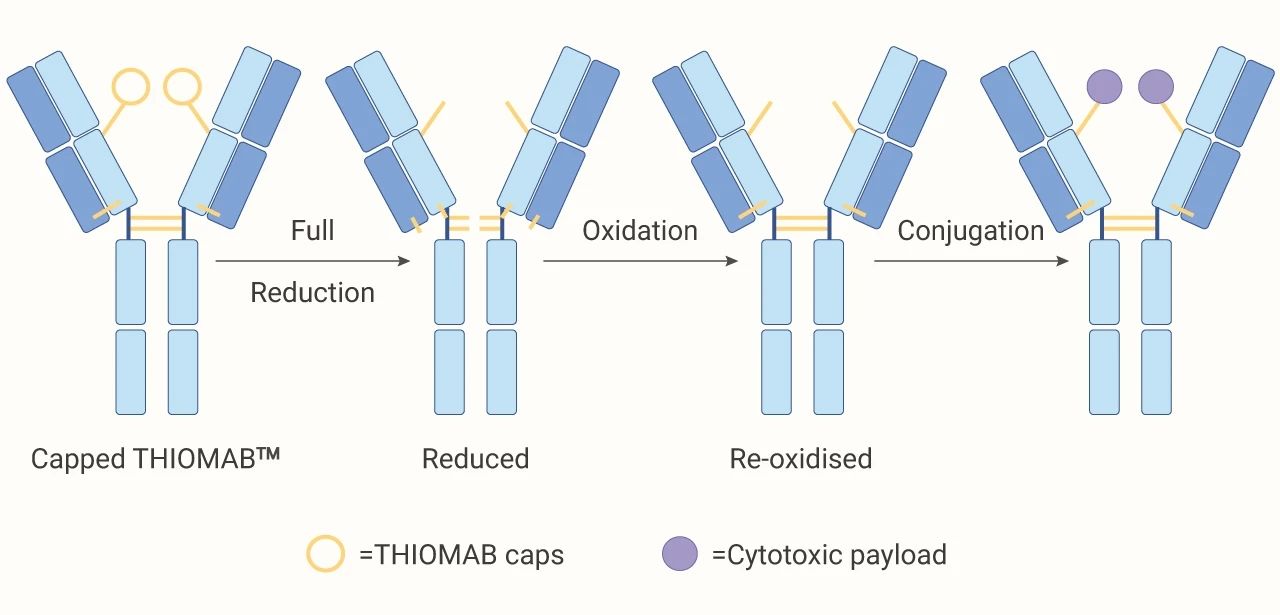
图 4. 位点特异性偶联过程
点击化学偶联:这种方法常常使用含叠氮化物的 linker,将叠氮化物基团连接到 payload 结构的末端。叠氮化物可以与加入抗体的 DBCO 基团反应。其优点是反应效率非常高和高产,条件温和。
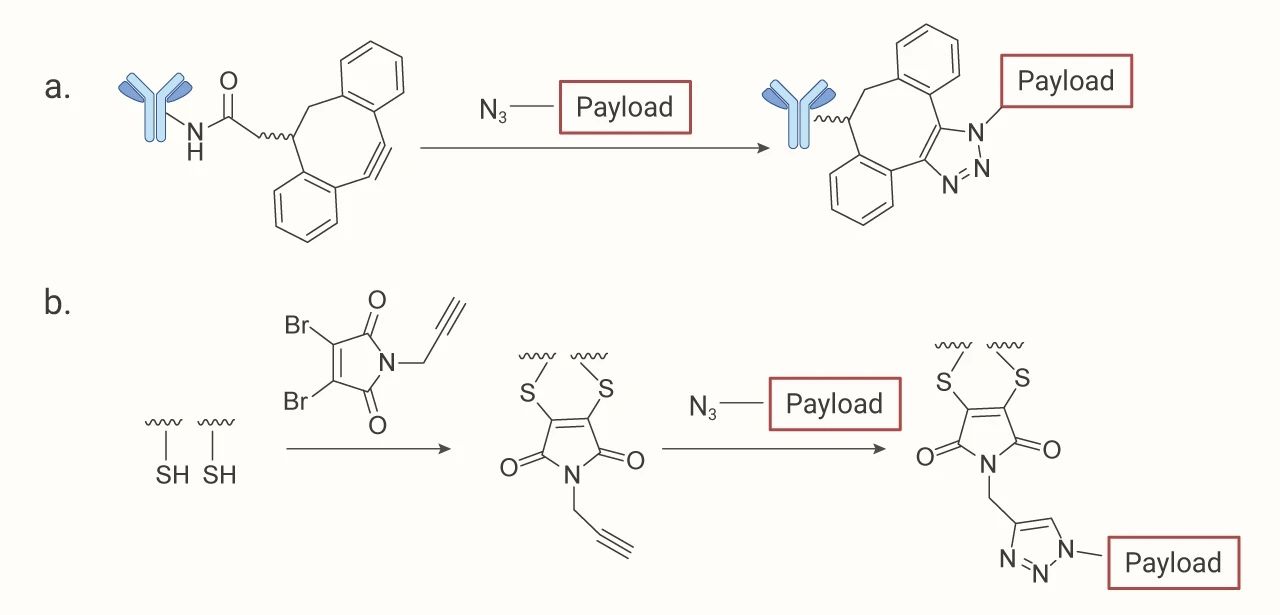
图 5. 点击化学偶联过程
其它偶联形式:除了上述以外,还有其它基于醇 (例如可以形成碳酸盐、醚和酯化合物) 和醛的偶联技术 (通过甲酰甘氨酸生成酶 (FGE) 偶联),通过工程氨基酰基——tRNA (aaRS)、氧化唾液酶和转氨试剂的偶联技术实现。
■ 那么如何检测 ADC 的“好与坏”呢?
药物抗体比 (DAR):目前已经发展了很多测定 DAR 的方案,包括全抗体质谱法、分光光度计测试法和各种色谱方法 (包括但不限于 HPLC、UV/VIS 紫外/可见光光谱分析)。
ADC 中未结合毒素的测定:反相 (RP)-HPLC 可用于根据外部标准曲线定量未缀合的有效载荷连接器的含量。
疏水性预测:在新 ADC 的发现和开发阶段,为了降低 ADC 的疏水性,尽可能降低有效毒素分子和连接子的分子量是至关重要的。在环结构和链中尽可能多的杂原子,以及尽可能多的亲水官能团作为取代基 (如-NH2、-OH/-COOH) 有助于优化亲水性。
ADC 的体外效价随着 DAR 的增加而增加,但是 ADC 的血浆清除率也随着 DAR 的增加而增加,从而减少了药物暴露和体内生物效果。亲水性改善后的 ADC 明显具有好的体内活性。亲水性改善后的ADC,血浆清除率降低,体内活性明显改善。
■ ADC 发展中的挑战和机遇
连接的不稳定性:这种不稳定性可导致 payload 过早释放到血液中,并导致 ADC 的非特异性摄取和脱靶毒性。一代的 ADC 具有酸可降解键 (如腙),其在偏中性 pH 的血浆中保持稳定,内化后在较低 pH 值的溶酶体中释放。但这种 linker (尤其是基于多肽的 linker) 会对血清蛋白酶敏感以及自发解偶联现象导致其在血浆中不稳定。和可降解 linker (SPP-DM1 和 VcMMAE) 相比,不可降解的 linker (mcc-DM1 和 mcMMAF) 具有更低的肝脏和血液毒性。因为其 payload 释放少,细胞毒性减小。但是不可降解 linker 不适用于所有靶点,毕竟需要单抗分解代谢才能释放 payload,另一方面,可降解 linker 导致 payload 的过早释放也可能通过“旁观者效应”发挥更好的疗效 (旁观者效应:活跃的 payload 也可能杀死周围的肿瘤细胞)。
非特异性内吞作用:一般来说,IgG 抗体的净正电荷增加导致血浆清除率增加,组织分布增加。因此,优化 ADC 表面电荷,减少正常细胞中非靶向摄取,同时保留靶点肿瘤细胞摄取,有利于改善治疗指标 (TI)。疏水性会促进 ADC、尤其是高 DAR 的 ADC 的聚集和非特异性内吞,从而产生脱靶效应。高 DAR 的 ADC 本身也会被其它具有非特异性、内吞能力强的细胞清除,因此优化 DAR 也是改善 TI 的重要策略。
受体介导的摄取机制:FcγRs 介导的 ADC 脱靶毒性主要体现在血液毒性。血液毒性是含有 Auristatin (MAME, MMAF)、Calicheamicin 和 Maytansinoid (DM-1) 的 ADC *常见的脱靶剂量限制毒性 dose-limiting toxicities (DLTs)。
| ADC |
| Trastuzumab emtansine 是一种抗体偶联药物 (ADC),其结合了 HER2 靶向的曲妥珠单抗的抗肿瘤特性以及微管抑制剂 DM1 的细胞毒活性。可用于晚期乳腺癌的研究。 |
| Trastuzumab deruxtecan 是一种抗人表皮生长因子受体 2 (HER2) 抗体-药物偶联物 (ADC)。由人源化抗 HER2 抗体,酶促裂解的肽接头和拓扑异构酶 I 抑制剂组成。可用于 HER2 阳性乳腺癌和胃癌的研究。 |
| ADC linker & Drug-Linker Conjugates for ADC |
| MC-Val-Cit-PAB 是一种可降解 (cleavable) 的 ADC linker,可用于合成抗体偶联药物 (ADC)。 |
| MC-Val-Cit-PAB-duocarmycin 是由 DNA 小沟结合烷化剂 duocarmycin 和 ADC linker MC-Val-Cit-PAB 连接而成,可用来制备抗体偶联药物。 |
| SMCC 是一种蛋白质交联剂。SMCC 接合抗原耦合脾脏细胞来诱导抗原特异性免疫反应。 |
| SMCC-DM1 是由微管破坏剂 DM1 和 ADC linker SMCC 连接而成,可用来制备抗体偶联药物。 |
| ADC Cytotoxin |
| Doxorubicin hydrochloride 一种具有细胞毒性的蒽环类抗生素,是一种抗癌化疗试剂。是一种有效的人类 DNA topoisomerase I 和 topoisomerase II 抑制剂,IC50 分别为 0.8 μM 和 2.67 μM。可降低 AMPK 及其下游靶蛋白乙酰辅酶 A 羧化酶的磷酸化。还可诱导凋亡 (apoptosis) 和自噬。 |
| Methotrexate 一种抗代谢 (antimetabolite) 和抗叶酸剂 (antifolate),可抑制二氢叶酸还原酶,从而防止叶酸转化为四氢叶酸并抑制 DNA 合成。也是一种免疫抑制剂和抗肿瘤剂,用于类风湿关节炎和研究多种癌症 (如急性淋巴细胞白血病)。 |
| Camptothecin 是有效的 DNA 拓扑异构酶 I 抑制剂,IC50 值为 679 nM。 |
| MMAF hydrochloride 是一种有效的微管蛋白聚合 (tubulin polymerization) 抑制剂,用作抗肿瘤药物。广泛用作抗体偶联药物 (ADCs) 的细胞毒性成分,如 Vorsetuzumab mafodotin 和 SGN-CD19A。 |
| Duocarmycin TM 是一种非常有效的有抗肿瘤活性的抗生素。是一种 DNA 烷化剂。 |
参考文献
1. Puregmaa Khongorzul, Juan Zhang, et al. Antibody-Drug Conjugates: A Comprehensive Review. Mol Cancer Res. 2020 Jan;18(1):3-19.
2. Ilona Pysz, Paul J. M. Jackson, David E. Thurston. Introduction to Antibody–Drug Conjugates (ADCs). Royal Society of Chemistry. 2019, pp. 1-30. Chapter 1.
3. Christina Peters, Stuart Brown. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015 Jun 12;35(4):e00225.
4. Xiuxia Sun, John M. Lambert, et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug Chem. 2017 May 17;28(5):1371-1381.
5. Aurijit Sarkar, Glen E. Kellogg. Hydrophobicity--shake flasks, protein folding and drug discovery. Curr Top Med Chem. 2010;10(1):67-83.
6. Robert P Lyon, Peter D Senter, et al. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015 Jul;33(7):733-5.
7. Prathap Kumar Mahalingaiah, et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol Ther. 2019 Aug;200:110-125
相关产品
-
 电议 型 号:
电议 型 号: -
电议 型 号:
-
电议 型 号:
-
电议 型 号:
-
电议 型 号:


